
La AHDS-MCT8
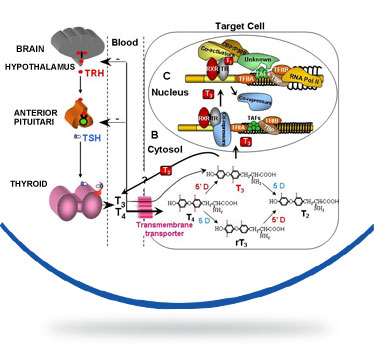
La sindrome di Allan-Herndon-Dudley (AHDS) o deficit del trasportatore degli ormoni tiroidei MCT8 è una rara encefalopatia congenita che colpisce i soggetti di sesso maschile caratterizzata da ritardo psicomotorio, ipotonia dell’asse, spasticità degli arti, distonia, epilessia. La malattia è dovuta a mutazioni del gene SLC16A2 che produce la proteina MCT8 che trasporta l’ormone tiroideo T3 all’interno delle cellule dove svolge le sue funzioni principali, in particolar modo per il corretto sviluppo del sistema nervoso. In questa malattia le concentrazioni di ormoni tiroidei all’interno nelle cellule nervose sono basse mentre le concentrazioni plasmatiche circolanti degli ormoni tiroidei (T3 e T4) sono alterate secondo un profilo molto particolare caratterizzato da un aumento del T3, di solito accompagnato da una riduzione del T4 e normali livelli dell’ormone ipofisario che stimola la produzione di ormoni tiroidei (TSH).
AHDS è una malattia ad ereditarietà X-linked recessiva che colpisce quindi solo i soggetti maschili. Con una stima di 14 pazienti in Italia e 200 nel mondo la sua prevalenza è inferiore ad 1 caso /1.000.000 di persone.. In linea generale i figli maschi di madri portatrici di mutazione su SLC16A2 hanno un rischio del 50% di essere affetti, mentre le femmine hanno il 50% di probabilità di essere a loro volta portatrici della mutazione La diagnosi prenatale di un maschio con AHDS è possibile se è nota la mutazione presente nella madre. Non esiste al momento una cura, il trattamento disponibile è solo sintomatico e di supporto.
In Italia è in corso un trial clinico sperimentale volto a valutare l’efficacia di una molecole che svolge funzione analoga al T3 ma che non necessita di MCT8 per entrare nelle cellule. Tale trial ha come centro coordinatore internazionale l’Erasmus Medical Center di Rotterdam (Dr. E. Visser, G Groenweg) e come centro coordinatore locale italiano l’Istituto Neurologico Carlo Besta di Milano (Dr.ssa F. Zibordi, Dr. D. Tonduti, Dr. N. Nardocci). In Italia partecipa al trial anche l’Ospedale Pediatrico Bambino Gesù di Roma (M. Dr. Cappa, Dr. E. Bertini)
English version
Allan–Herndon–Dudley Syndrome is a rare X-linked inherited congenital hypothyroidism that causes characterized by hypoplasia ( abnormal development of an organ or tissue ) and severe psychomotor retardation. This condition, which occurs almost exclusively in males, disrupts development from before birth.
Allan–Herndon–Dudley syndrome, which is named eponymously for William Allan, Florence C. Dudley, and C. Nash Herndon,[1][2] results from a mutation of the thyroid hormone transporter MCT8 (also referred to as SLC16A2). Consecutively, thyroid hormones are unable to enter the nervous system, which depends on thyroid signaling for proper function and development.
The disease is caused by a mutation of the gene SLC16A2 or MCT8 that produces a protein that carries T3 thyroid hormone to the central nervous system . This hormone is essential for the proper development of neurons and their synapses. The absence or alteration of this protein does not allow the transport of the hormone T3 to the central nervous system , and consequently the development of nerve cells is interrupted .
The hormone is not transported to the central nervous system so it creates an excess of T3 in the blood , which goes to accumulate in other organs , causing additional signs and symptoms . The serum parameters of thyroid hormones are highly abnormal in AHDS syndrome: there is an high T3 value, a low T4 and normal TSH level. High blood level of thyroid hormone T3 gives as result a low body weight and a reduced muscle mass.
Most children with Allan–Herndon–Dudley syndrome have weak muscle tone (hypotonia) and underdevelopment of many muscles (muscle hypoplasia). As they get older, they usually develop joint deformities called contractures, which restrict the movement of certain joints. Abnormal muscle stiffness (spasticity), muscle weakness, and involuntary movements of the arms and legs also limit mobility. As a result, many people with Allan–Herndon–Dudley syndrome are unable to walk independently and become wheelchair-bound by adulthood.
Mutations in the SLC16A2 gene cause Allan–Herndon–Dudley syndrome. The SLC16A2 gene, also known as MCT8, provides instructions for making a protein that plays a critical role in the development of the nervous system. This protein transports a particular hormone into nerve cells in the developing brain. This hormone, called triiodothyronine or T3, is produced by the thyroid. T3 appears to be critical for the normal formation and growth of nerve cells, as well as the development of junctions between nerve cells (synapses) where cell-to-cell communication occurs. T3 and other forms of thyroid hormone also help regulate the development of other organs and control the rate of chemical reactions in the body.
Gene mutations alter the structure and function of the SLC16A2 protein. As a result, this protein is unable to transport T3 into nerve cells effectively. A lack of this critical hormone in certain parts of the brain disrupts normal brain development, resulting in intellectual disability and problems with movement. Excess amounts of T3 circulate in the bloodstream. Increased T3 levels in the blood may be toxic to some organs and contribute to the signs and symptoms of Allan–Herndon–Dudley syndrome.
The disease is inherited by X -linked recessive mode and its incident is 1 / 1,000,000 . To affected families should be offered genetic counseling and should be informed that the males have a 50% risk of being affected if the mother is a carrier of a mutation in SLC16A2 , while females have a 50 % chance of inheriting the mutation if their mother is heterozygous for the mutation. Prenatal diagnosis of a male with AHDS is possible if the mother is a carrier of a specific mutation in SLC16A2.
There is currently no cure, treatment is symptomatic and supportive.
In the Netherlands at the Erasmus Medical Center in Rotterdam a medical trial is currently on the way where an hormone substitute similar to T3 that is not dependent to MCT8 for its entry into cells is given to patients.
This hormone substitute called Triac (triiodothyroacetate or Tiratricol, a natural thyroid hormone) may at least partially restore the abnormalities found in patients with AHDS. Some studies in vivo, in vitro and in animals have shown that the Triac is a very promising candidate to fulfill this function.
The Triac binds to the same receptors of thyroid hormones in the same manner of T3; cellular uptake of the Triac does not depend on correct function of MCT8 that in patients suffering from this disease doesn’t work, therefore, in patients suffering from AHDS, the Triac can be provided also in the tissues that require that MCT8 is working for absorption of the thyroid hormones themselves, as for example, the brain.
In vitro studies have shown that neuronal cells differentiate equally well in the presence either of Triac or T3; In Mct8 deficient mice, Triac is taken up by the brain and suppresses serum TSH levels; consequently, serum T3 and T4 levels were lowered; Triac is the treatment of choice in patient with the resistance to thyroid hormone (RTH) syndrome. Patient with RTH have high serum TSH and thyroid hormone levels, which shows strong similarities to the profile found in AHDS patients; the longstanding experience with Triac in RTH indicates its safety and tolerability .
Thus, Triac treatment could result in normalization of the abnormal serum Thyroid hormones values in AHDS patients. Furthermore, Triac could replace the function of T3 in tissues that depend on MCT8 for Thyroid hormones uptake (e.g. brain).
The current trial in the Netherlands is responsible to investigate whether treatment with the Triac in patients with ADHS will reduce the toxic effects of high levels T3 and can restore the lack of thyroid hormones in the brain.

In Programma
Non ci sono eventi in arrivo al momento.